

هیپر فنیل آلانینمی می تواند در اثر وجود نقص در ژن های سازنده کوفاکتور تتراهیدروبیوپترین نیز بروز پیدا می کند که اولین بیمار با این شرایط در سال 1970 گزارش شد. شیوع فنیل کتونوریا حاصل از نقص در سنتز BH4 1 در 1000000 تولد تخمین زده شده است اما بر حسب مناطق جغرافیایی مختلف این اندازه متفاوت است. بعنوان مثال در بخشی از ایتالیا حدود 10% بیماران هیپر فنیل آلانینمی، در ترکیه 15%، در تایوان 19% و در عربستان صعودی حدود 68% را تشکیل می دهد.
ژنتیک و علل ایجاد کننده بیماری:
حدود 2% از بیماران هیپر فنیل آلانینمی حاصل از جهش در یکی از ژن های مذکور در سنتز BH4(همگی دارای توارث اتوزومال مغلوب)
یافته های بالینی:
نوزادان مبتلا دچار کاهش وزن، هیپوتونی و هیپرتونی در دست و پا، در 2-3 ماهگی کاهش فعالیت یا از دست دادن کنترل سر، اختلالات عصبی، برادی کینزی[1]، کجی گردن[2]، اپیستوتونیک، دست های خمیده[3]، کلونوس[4]، به سختی غذا خوردن، بی حالی[5]، کج خلقی[6]، اسپاسم های چشمی[7]، رعشه[8]، تشنج میوکلونیک[9]، میکروسفالی غدد عصبی بازال[10] غیر طبیعی، کلسیفیکیشن[11] در مغز مشاهده می شوند. نقص در سنتز نوروترانسمیترها باعث افزایش اختلالات تکوینی و عصبی در این بیماران می گردد.
بیماریزایی:
سنتز کوفاکتور تتراهیدروبیوپترین از گوانوزین تری فسفات (GTP) صورت می گیرد که از چندین مرحله احیا نئوپترین (8،7 دی هیدرونئوپترین تری فسفات) که یک میانکنش است، تشکیل شده است. مراحل آن به ترتیب بوسیله آنزیـم هـای GTP سیکلـوهیـدراتــازI ،[12](GTPCH) 6-پیروات تتراهیدروپترین سنتتاز[13](6-PTS)، سپیاپتریـن ردوکتـاز[14]، آلـدوز ردوکتـاز، کربونیل ردوکتاز انجام می پذیرد. نقص در آنزیم سپیاپترین ردوکتاز هنوز با هیچ بیماری انسانی تا به حال مطرح نشده است. فراوانی نقص آنزیمی در بیماران غیر کلاسیک فنیل کتونوریا به ترتیب عبارتند از نقـص در 6-PTS، DHFR، PCD، GTPCH.

برخی از این اختلالات در این دستـه قـرار نمی گیرند به عنوان مثال در بیماری ساگوا [15] یا دی هیدروکسی فنیل آلانین دیستونی[16] که در نتیجه جهش های اتوزومال غالب در ژن GTPCH ایجاد می گردد هیپر فنیل آلانین دیده نمی شود. 60% بیماران فنیل کتونوریا غیر کلاسیک به علت نقص در 6-PTS به این بیماری مبتلا شده اند. آنالیزcDNA حاصل از 6 اگزون ژن 6-PTS که شامل 435 bp می باشد، 28 موتاسیون تاکنون شناسایی شده است که دو تا از این جهش ها از نوع جهش های جایگاه برش می باشند. سایر جهش ها از انواع حذف، نقطـه ای و کد پایانی می باشند. شیوع بالا جهش های N52S و P87Sدر بیماران آسیایی وجود دارد. در بیمارانی با ژنوتیپ هموزیگوت جهش I114V: هیپر فنیل آلانینمی ملایم و دیستونی ایجاد می کند.
در 4 درصد این بیماران نقص در GTPCH دیده شده است. ژن GTPCH دارای 6 اگزون می باشد. سه جهش اعم از M211I، R184H و Q110X در یک سوم بیماران نقص در DHPR دیده می شوند.
ژن DHPR کد کننده پروتئینی 25.7 kD می باشد. یک سری جهش در سرتاسر این ژن پراکنده شده است. وارد شدن یک اسید آمینه ترئونین در میان آلانین 122 و ترئونین 123، جهش نقطه ای T122A
در 4 درصد بیماران نقص در ژن PCD که کد کننده یک فاکتور رونویسی است و دارای 4 اگزون (بیش از 5kb) وجود دارد. دارای جهش های زیاد بی معنایی که اکثرا در اگزون 4 است.
تتراهیدروبیوپترین کوفاکتور برای هیدروکسیلاسیون اسید آمینه های تریپتوفان و تیروزین نیز می باشد که موجب نقص در تولید سرتونین[17]، دی هیدروکسی فنیل آلانین[18] (DOPA) و نورپین فرین[19] می گردد.
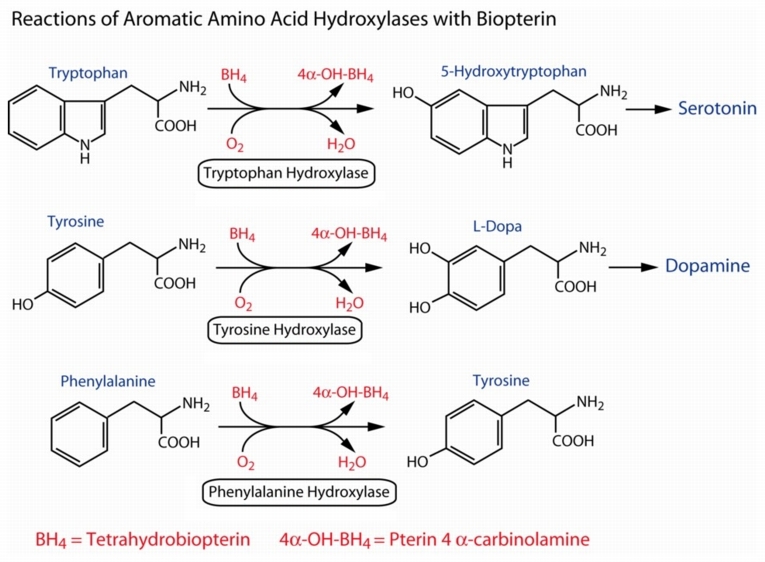
راه های تشخیصی:
سطح پایین قابل توجه 5-هیدروکسی ایندول استیک اسید[20]، ونیلیل ماندلیک اسید[21] و همووانیلیک اسید[22] در ادرار و مایع مغزی نخاعی و همچنین سطح کاهش یافته دوپامین و سرتونین در ادرار می تواند راه تشخیصی مناسبی باشند. نقص در سنتز نوروترانسمیترها باعث افزایش اختلالات تکوینی و عصبی در این بیماران می گردد. بیمارانی که دارای نقص در یکی از آنزیم های مسیر متابولیکی BH4 در زمان تولد کاملا طبیعی می باشند ولی در تست غربالگری غلظت افزایش یافته فنیل آلانین در خون را نشان می دهند.
مشاوره ژنتیک:
در صورت عدم درمان در پنج سال اول زندگی می میرند. در مورد بیماران مبتلا به فنیل کتونوریا که دارای نقص در سنتز BH4 هستند، رژیم غذایی با کاهش فنیل آلانین در مورد آن ها مناسب نمی باشد.
خانواده های دارای فرد مبتلا باید مورد مشاوره ژنتیک قرار گیرند و در صورت لزوم تست ناقلیت، تست تشخیص قبل از تولد برای افراد در معرض خطر خانواده انجام پذیرد.
[1] Bradykinesia
[2] Torticollis
[3] Pronate
[4] Clonus
[5] Expressionless
[6] Irritability
[7] Oculogyric spasms
[8] Tremors
[9] Myoclonic siezure
[10] Basal ganglia
[11] Calcification
[12] GTPcyclohydrolase I
[13] 6-pyruvoyltetrahydropterin synthase
[14] Sepiapterin reductase
[15]Sagwa
[16] Dystonia
[17] Serotonin
[18] Dihydroxyphenylalanine
[19] Norepinephrine
[20] 5-hydroxyindoleacetic acid
[21] Vanillylmandelic acid
[22] Homovanillic acid
برچسب های مهم
اگر به یک وب سایت یا فروشگاه رایگان با فضای نامحدود و امکانات فراوان نیاز دارید بی درنگ دکمه زیر را کلیک نمایید.
ایجاد وب سایت یا Movable DNA
Movable DNA Molecular Cell Biology
Molecular Cell Biology Possum
Possum DNA
DNA Colorectal Cancer Screening Quality and Benchmarks
Colorectal Cancer Screening Quality and Benchmarks Silver DNA
Silver DNA Comprehensive Textbook of Hepatitis B
Comprehensive Textbook of Hepatitis B Parental Genetic Testing
Parental Genetic Testing THOMPSON & THOMPSON GENETICS IN MEDICINE 8th
THOMPSON & THOMPSON GENETICS IN MEDICINE 8th Atlas of Metabolic Diseases
Atlas of Metabolic Diseases.png) وراثت اتوزومال غالب
وراثت اتوزومال غالب هتروژنی لوکوسی
هتروژنی لوکوسی Chromosome translocations
Chromosome translocations Hypochondroplasia
Hypochondroplasia هتروژنی آللی
هتروژنی آللی Achondroplasia
Achondroplasia ژن مو قرمز با خطری برابر دو دهه در مقابل نور خورشید قرار گرفتن می تواند موجب ایجاد جهش های سرطان زا پوست گردد
ژن مو قرمز با خطری برابر دو دهه در مقابل نور خورشید قرار گرفتن می تواند موجب ایجاد جهش های سرطان زا پوست گردد Blepharophimosis – Ptosis – Epicanthus Inversus Syndrome
Blepharophimosis – Ptosis – Epicanthus Inversus Syndrome Phenylketonuria
Phenylketonuria Klippel Feil Syndrome
Klippel Feil Syndrome Movable DNA
Movable DNA Circular DNA
Circular DNA Silver DNA
Silver DNA CELL FREE FETAL DNA
CELL FREE FETAL DNA Genetics and Genomics in Medicine
Genetics and Genomics in Medicine Alzheimers disease & DNA
Alzheimers disease & DNA Possum
Possum Atlas of Metabolic Diseases
Atlas of Metabolic Diseases Atlas of Genetic Diagnosis and Counseling
Atlas of Genetic Diagnosis and Counseling Chromosome Abnormalities and Genetic Counseling Movable DNA DNA Possum Molecular Cell Biology
Chromosome Abnormalities and Genetic Counseling Movable DNA DNA Possum Molecular Cell Biology Alzheimers disease & DNA
Alzheimers disease & DNA CELL FREE FETAL DNA Silver DNA
CELL FREE FETAL DNA Silver DNA Molecular Biology of Cancer Mechanisms, Targets, and Therapeutics THOMPSON & THOMPSON GENETICS IN MEDICINE 8th
Molecular Biology of Cancer Mechanisms, Targets, and Therapeutics THOMPSON & THOMPSON GENETICS IN MEDICINE 8th Essential Medical Genetics
Essential Medical Genetics Notch Signaling in Embryology and Cancer
Notch Signaling in Embryology and Cancer Genetic Counseling Research: A Practical Guide
Genetic Counseling Research: A Practical Guide







 دانلود جزوه پاتولوژی عمومی
دانلود جزوه پاتولوژی عمومی دانلود گیاهان دارویی علی زرگری 5 جلدی pdf
دانلود گیاهان دارویی علی زرگری 5 جلدی pdf گزارش کاراموزی ترم 7 و 8 رادیولوژِی(MRI,سی تی اسکن و عکس رنگی)
گزارش کاراموزی ترم 7 و 8 رادیولوژِی(MRI,سی تی اسکن و عکس رنگی) مقاله آناتومی و فیزیولوژی
مقاله آناتومی و فیزیولوژی دانلود کتاب ایمونولوژی رویت ترجمه فارسی
دانلود کتاب ایمونولوژی رویت ترجمه فارسی